



1 LÄKEMEDLETS NAMN
Inlyta 1 mg filmdragerade tabletter
Inlyta 3 mg filmdragerade tabletter
Inlyta 5 mg filmdragerade tabletter
Inlyta 7 mg filmdragerade tabletter
2 KVALITATIV OCH KVANTITATIV SAMMANSÄTTNING
Inlyta 1 mg filmdragerade tabletter
Varje filmdragerad tablett innehåller 1 mg axitinib.
Inlyta 3 mg filmdragerade tabletter
Varje filmdragerad tablett innehåller 3 mg axitinib.
Inlyta 5 mg filmdragerade tabletter
Varje filmdragerad tablett innehåller 5 mg axitinib.
Inlyta 7 mg filmdragerade tabletter
Varje filmdragerad tablett innehåller 7 mg axitinib.
Hjälpämnen med känd effekt:
Inlyta 1 mg filmdragerad tablett
Varje filmdragerad tablett innehåller 33,6 mg laktosmonohydrat.
Inlyta 3 mg filmdragerad tablett
Varje filmdragerad tablett innehåller 35,3 mg laktosmonohydrat.
Inlyta 5 mg filmdragerad tablett
Varje filmdragerad tablett innehåller 58,8 mg laktosmonohydrat.
Inlyta 7 mg filmdragerad tablett
Varje filmdragerad tablett innehåller 82,3 mg laktosmonohydrat.
För fullständig förteckning över hjälpämnen, se avsnitt 6.1.
3 LÄKEMEDELSFORM
Filmdragerad tablett (tablett).
Inlyta 1 mg filmdragerade tabletter
Röd oval filmdragerad tablett med ”Pfizer” präglat på den ena sidan och ”1 XNB” på den andra.
Inlyta 3 mg filmdragerade tabletter
Röd rund filmdragerad tablett med ”Pfizer” präglat på den ena sidan och ”3 XNB” på den andra.
Inlyta 5 mg filmdragerade tabletter
Röd triangelformad filmdragerad tablett med ”Pfizer” präglat på den ena sidan och ”5 XNB” på den andra.
Inlyta 7 mg filmdragerade tabletter
Röd rombformad filmdragerad tablett med ”Pfizer” präglat på den ena sidan och ”7 XNB” på den andra.
4 KLINISKA UPPGIFTER
4.1 Terapeutiska indikationer
Inlyta är indicerat för behandling av vuxna patienter med avancerad njurcancer (RCC) efter svikt på tidigare behandling med sunitinib eller cytokin.
4.2 Dosering och administreringssätt
Behandling med Inlyta ska utföras av läkare med erfarenhet av cancerbehandling.
Dosering
Rekommenderad dos av axitinib är 5 mg två gånger dagligen.
Behandlingen ska fortsätta så länge klinisk nytta observeras eller tills oacceptabel toxicitet inträffar, som inte kan behandlas genom samtidig läkemedelsbehandling eller genom dosjustering.
Om patienten kräks eller missar en dos ska ingen extra dos tas. Nästa ordinerade dos ska tas vid vanlig tidpunkt.
Dosjusteringar
Dosökning eller dosminskning rekommenderas baserat på säkerhet och tolerabilitet för den enskilde patienten.
Patienter som tolererar startdosen av axitinib, 5 mg två gånger dagligen, utan biverkningar > grad 2 (dvs. utan svåra biverkningar enligt Common Terminology Criteria for Adverse Events (CTCAE) version 3.0) under två veckor i följd, kan få ökad dos. Den kan höjas till 7 mg två gånger dagligen om patientens blodtryck inte överstiger 150/90 mmHg eller patienten får blodtryckssänkande behandling. Med samma kriterier som grund kan patienter som tolererar axitinib i dosen 7 mg två gånger dagligen få dosen ökad till maximalt 10 mg två gånger dagligen.
För att kunna hantera vissa biverkningar kan behandlingen behöva avbrytas tillfälligt eller permanent, och/eller kan axitinibdosen behöva sänkas (se avsnitt 4.4). Om dossänkning krävs kan dosen axitinib sänkas till 3 mg två gånger dagligen och ytterligare till 2 mg två gånger dagligen.
Dossänkning baserat på patientens ålder, etnicitet, kön eller kroppsvikt krävs inte.
Samtidig behandling med starka CYP3A4/5-hämmare
Samtidig administrering av axitinib och starka CYP3A4/5-hämmare kan öka plasmakoncentrationen av axitinib (se avsnitt 4.5). Alternativt läkemedel som saknar, eller har minimal, hämmande effekt på CYP3A4/5 rekommenderas.
Justering av axitinibdosen till patienter som får starka CYP3A4/5-hämmare har inte studerats, men om en stark CYP3A4/5-hämmare måste ges samtidigt rekommenderas en sänkning av axitinibdosen till ungefär halva dosen (dvs. startdosen bör sänkas från 5 mg två gånger dagligen till 2 mg två gånger dagligen). För att kunna hantera vissa biverkningar kan axitinibbehandlingen behöva avbrytas tillfälligt eller permanent (se avsnitt 4.4). Om samtidig administrering av den starka hämmaren avbryts, ska återgång till den axitinibdos som användes före behandlingen med den starka CYP3A4/5-hämmaren övervägas (se avsnitt 4.5).
Samtidig behandling med starka CYP3A4/5-inducerare
Samtidig administrering av axitinib och starka CYP3A4/5-inducerare kan sänka plasmakoncentrationen av axitinib (se avsnitt 4.5). Alternativt läkemedel som saknar, eller har minimal, CYP3A4/5-inducerande effekt rekommenderas.
Justering av axitinibdosen till patienter som får starka CYP3A4/5-inducerare har inte studerats, men om en stark CYP3A4/5-inducerare måste ges samtidigt rekommenderas gradvis ökning av axitinibdosen. Maximal induktion med starka CYP3A4/5-inducerare i höga doser har rapporterats inträffa inom en vecka efter att behandlingen med induceraren inleddes. Om axitinibdosen höjs ska patienten övervakas noga avseende toxicitet. För att kunna hantera vissa biverkningar kan axitinibbehandlingen behöva avbrytas tillfälligt eller permanent, och/eller kan dosen axitinib behöva sänkas (se avsnitt 4.4). Om samtidig administrering av den starka induceraren avbryts, ska den axitinibdos som användes före behandlingen med den starka CYP3A4/5-induceraren omedelbart återupptas (se avsnitt 4.5).
Särskilda populationer
Äldre (≥ 65 år)
Ingen dosjustering behövs (se avsnitt 4.4 och 5.2).
Nedsatt njurfunktion
Ingen dosjustering behövs (se avsnitt 5.2). Det finns praktiskt taget inga data om axitinibbehandling till patienter med kreatininclearance < 15 ml/min.
Nedsatt leverfunktion
Ingen dosjustering behövs när axitinib ges till patienter med lätt nedsatt leverfunktion (Child-Pugh klass A). Dosminskning rekommenderas när axitinib ges till patienter med måttligt nedsatt leverfunktion (Child-Pugh klass B) (dvs. startdosen bör sänkas från 5 mg två gånger dagligen till 2 mg två gånger dagligen). Axitinib har inte studerats hos patienter med allvarligt nedsatt leverfunktion (Child-Pugh klass C) och bör inte användas i denna population (se avsnitt 4.4 och 5.2).
Pediatrisk population
Säkerhet och effekt för Inlyta för barn och ungdomar < 18 år har inte fastställts. Inga data finns tillgängliga.
Administreringssätt
Axitinib är för oral användning. Tabletterna ska tas peroralt två gånger dagligen med cirka 12 timmars intervall, med eller utan föda (se avsnitt 5.2). De ska sväljas hela med ett glas vatten.
4.3 Kontraindikationer
Överkänslighet mot axitinib eller mot något hjälpämne som anges i avsnitt 6.1.
4.4 Varningar och försiktighet
Specifika händelser som gäller säkerheten ska följas upp före, och med jämna mellanrum under, behandlingen med axitinib enligt beskrivning här nedan.
Hjärtsviktshändelser
I kliniska studier av axitinib för behandling av patienter med RCC rapporterades hjärtsviktshändelser, däribland hjärtsvikt, kongestiv hjärtsvikt, hjärtlungsvikt, vänsterkammardysfunktion, minskad ejektionsfraktion samt högerkammarsvikt (se avsnitt 4.8).
Tecken och symtom på hjärtsvikt ska övervakas regelbundet under hela behandlingen med axitinib. För att hantera hjärtsviktshändelser kan det bli nödvändigt med tillfälligt avbrott eller permanent utsättning av behandlingen med axitinib, och/eller dosreduktion.
Hypertoni
I kliniska studier av axitinib för behandling av patienter med RCC var hypertoni en mycket vanlig biverkan (se avsnitt 4.8).
I en kontrollerad klinisk studie debuterade hypertonin (systoliskt blodtryck > 150 mmHg eller diastoliskt blodtryck > 100 mmHg) i median inom en månad efter att behandlingen med axitinib inletts. Förhöjt blodtryck har observerats redan 4 dagar efter behandlingsstarten med axitinib.
Blodtrycket bör vara väl kontrollerat innan behandling med axitinib inleds. Patienternas blodtryck bör följas och behandlas efter behov med rutinmässig blodtryckssänkande behandling. Vid kvarstående hypertoni trots blodtryckssänkande läkemedel bör axitinibdosen sänkas. För patienter som utvecklar svår hypertoni bör axitinibbehandlingen avbrytas tillfälligt och återupptas vid en lägre dos när blodtrycket har normaliserats. Om behandlingen med axitinib avbryts bör patienter som får blodtryckssänkande läkemedel kontrolleras för hypotoni (se avsnitt 4.2).
Vid svår eller kvarstående arteriell hypertoni och symtom som tyder på posteriort reversibelt encefalopatisyndrom (PRES) (se nedan) bör en diagnostisk magnetkameraundersökning (MRT) av hjärnan övervägas.
Rubbningar i sköldkörtelfunktionen
I kliniska studier av axitinib för behandling av patienter med RCC rapporterades hypotyreos och, i mindre omfattning, hypertyreos (se avsnitt 4.8).
Sköldkörtelfunktionen bör kontrolleras innan behandling med axitinib inleds och med jämna mellanrum under behandlingen. Hypo- eller hypertyreos bör behandlas enligt normal medicinsk praxis för att bibehålla normal sköldkörtelfunktion.
Arteriella emboliska och trombotiska händelser
I kliniska studier av axitinib rapporterades arteriella emboliska och trombotiska händelser (däribland transitorisk ischemisk attack (TIA), hjärtinfarkt, cerebrovaskulär händelse och näthinneartärsocklusion) (se avsnitt 4.8).
Axitinib bör användas med försiktighet av patienter som löper risk för, eller tidigare drabbats av, sådana händelser. Axitinib har inte studerats hos patienter som haft en arteriell embolisk eller trombotisk händelse under de senaste 12 månaderna.
Venösa emboliska och trombotiska händelser
I kliniska studier av axitinib rapporterades venösa emboliska och trombotiska händelser (däribland lungemboli, djup ventrombos och näthinnevensocklusion/-trombos) (se avsnitt 4.8).
Axitinib bör användas med försiktighet till patienter som löper risk för, eller tidigare drabbats av, sådana händelser. Axitinib har inte studerats hos patienter som haft en venös embolisk eller trombotisk händelse under de senaste 6 månaderna.
Förhöjt hemoglobin- eller hematokritvärde
Förhöjda hemoglobin- eller hematokritvärden, som återspeglar en ökning av mängden röda blodkroppar, kan uppträda under behandling med axitinib (se avsnitt 4.8 Polycytemi). En ökad mängd röda blodkroppar kan öka risken för emboliska och trombotiska händelser.
Hemoglobin- eller hematokritvärdena bör kontrolleras innan behandling med axitinib inleds och med jämna mellanrum under behandlingen. Om hemoglobin- eller hematokritvärdet stiger över normalvärdet bör patienten behandlas enligt medicinsk praxis för att sänka hemoglobin- eller hematokritvärdet till en godtagbar nivå.
Blödning
Blödning har rapporterats i kliniska studier av axitinib (se avsnitt 4.8).
Axitinib har inte studerats hos patienter med obehandlade hjärnmetastaser eller nyligen inträffad gastrointestinal blödning och bör inte användas till dessa patienter. Om en blödning kräver medicinsk intervention bör axitinibdosen tillfälligt sättas ut.
Aneurysmer och arteriella dissektioner
Användningen av VEGF-hämmare till patienter med eller utan hypertoni kan främja bildningen av aneurysmer och/eller arteriella dissektioner. Denna risk ska noga övervägas innan Inlyta sätts in hos patienter med riskfaktorer såsom hypertoni eller tidigare aneurysm.
Gastrointestinal perforation och fistelbildning
Gastrointestinal perforation och fistelbildning har rapporterats i kliniska studier av axitinib (se avsnitt 4.8).
Symtom på gastrointestinal perforation eller fistelbildning bör kontrolleras regelbundet under behandlingen med axitinib.
Sårläkningskomplikationer
Inga formella studier av axitinibs effekter på sårläkning har genomförts.
Behandlingen med axitinib bör avbrytas minst 24 timmar före ett planerat kirurgiskt ingrepp. Beslutet om att återuppta axitinibbehandlingen efter ingreppet bör baseras på en klinisk bedömning av sårläkningsprocessen.
Posteriort reversibelt encefalopatisyndrom (PRES)
Posteriort reversibelt encefalopatisyndrom (PRES) har rapporterats i kliniska studier av axitinib (se avsnitt 4.8).
PRES är ett neurologiskt tillstånd som kan visa sig som huvudvärk, kramper, letargi, förvirring, blindhet och andra synrubbningar och neurologiska rubbningar. Lätt till svår hypertoni kan förekomma. Magnetresonanstomografi krävs för att bekräfta diagnosen PRES. Om patienten visar tecken eller symtom på PRES bör behandlingen med axitinib avbrytas tillfälligt eller permanent. Säkerheten vid återinsättning av axitinibbehandling till patienter med tidigare PRES är inte känd.
Proteinuri
Proteinuri, inklusive av svårighetsgrad 3 och 4, har rapporterats i kliniska studier av axitinib (se avsnitt 4.8).
Kontroller avseende proteinuri rekommenderas innan behandling med axitinib inleds och med jämna mellanrum under behandlingen. Om patienten utvecklar måttlig till svår proteinuri bör dosen sänkas eller behandlingen tillfälligt avbrytas (se avsnitt 4.2). Behandling med axitinib ska avbrytas om patienten utvecklar nefrotiskt syndrom.
Leverrelaterade biverkningar
I en kontrollerad klinisk studie av axitinib för behandling av patienter med RCC rapporterades leverrelaterade biverkningar. De vanligaste leverrelaterade biverkningarna var ökningar av ALAT- (alaninaminotransferas), ASAT- (aspartataminotransferas) och bilirubinvärdena (se avsnitt 4.8). Inga samtidiga höjningar av ALAT (> 3 x övre normalvärdet (ULN)) och bilirubin (> 2 x ULN) observerades.
I en klinisk dosundersökningsstudie observerades samtidig höjning av ALAT (12 x ULN) och bilirubin (2,3 x ULN), vilket bedömdes som en läkemedelsrelaterad levertoxicitet, hos en patient som fick axitinib med startdosen 20 mg två gånger dagligen (4 gånger den rekommenderade startdosen).
Leverfunktionen bör kontrolleras innan behandling med axitinib inleds och med jämna mellanrum under behandlingen.
Nedsatt leverfunktion
I kliniska studier av axitinib var systemexponeringen för axitinib ungefär dubbelt så hög hos patienter med måttligt nedsatt leverfunktion (Child-Pugh klass B) som hos patienter med normal leverfunktion. Dosminskning rekommenderas när axitinib ges till patienter med måttligt nedsatt leverfunktion (Child-Pugh klass B) (se avsnitt 4.2).
Axitinib har inte studerats hos patienter med allvarligt nedsatt leverfunktion (Child-Pugh klass C) och bör inte användas i denna population.
Äldre (≥ 65 år) och etnicitet
I en kontrollerad klinisk studie av axitinib för behandling av patienter med RCC var 34 % av de behandlade patienterna ≥ 65 år. Majoriteten av patienterna var ljushyade (77 %) eller asiatiska (21 %). Även om man inte kan utesluta en större benägenhet hos en del äldre patienter och asiatiska patienter att utveckla biverkningar, observerades inga större skillnader i säkerhet och effekt för axitinib mellan de patienter som var ≥ 65 år och de yngre patienterna, eller mellan ljushyade och andra etniciteter.
Ingen dosjustering krävs med hänsyn till patientens ålder eller etnicitet (se avsnitt 4.2 och 5.2).
Hjälpämnen
Laktos
Detta läkemedel innehåller laktos. Patienter med något av följande sällsynta ärftliga tillstånd bör inte använda detta läkemedel: galaktosintolerans, total laktasbrist eller glukos-galaktosmalabsorption.
Natrium
Detta läkemedel innehåller mindre än 1 mmol (23 mg) natrium per filmdragerad tablett, d.v.s. är näst intill ”natriumfritt”.
4.5 Interaktioner med andra läkemedel och övriga interaktioner
In vitro-data visar att axitinib främst metaboliseras av CYP3A4/5, och i mindre utsträckning av CYP1A2, CYP2C19 och uridindifosfat-glukuronosyltransferas (UGT) 1A1.
CYP3A4/5-hämmare
Ketokonazol, en stark CYP3A4/5-hämmare, administrerad i en dos om 400 mg en gång dagligen i 7 dagar, fördubblade genomsnittlig area under kurvan (AUC) och ökade Cmax 1,5 gånger för en peroral singeldos om 5 mg axitinib hos friska frivilliga försökspersoner. Samtidig administrering av axitinib och starka CYP3A4/5-hämmare (t.ex. ketokonazol, itrakonazol, klaritromycin, erytromycin, atazanavir, indinavir, nefazodon, nelfinavir, ritonavir, sakvinavir och telitromycin) kan öka plasmakoncentrationen av axitinib. Grapefrukt kan också öka plasmakoncentrationen av axitinib. Vid val av läkemedel för samtidig administrering rekommenderas sådana utan eller med minimal hämmande effekt på CYP3A4/5. Om en stark CYP3A4/5-hämmare måste ges samtidigt rekommenderas dosjustering av axitinib (se avsnitt 4.2).
CYP1A2- och CYP2C19-hämmare
CYP1A2 och CYP2C19 utgör mindre (< 10 %) elimineringsvägar för axitinib. Effekten på axitinibs farmakokinetik vid tillförsel av starka hämmare av dessa isoenzymer har inte studerats. Försiktighet bör iakttas på grund av risken för förhöjda plasmakoncentrationer av axitinib hos patienter som tar starka hämmare av dessa isoenzymer.
CYP3A4/5-inducerare
Rifampicin, en stark CYP3A4/5-inducerare, administrerad i en dos om 600 mg en gång dagligen i 9 dagar, minskade genomsnittlig area under kurvan (AUC) med 79 % och Cmax med 71 % för en singeldos om 5 mg axitinib som gavs friska frivilliga försökspersoner.
Samtidig administrering av axitinib och starka CYP3A4/5-inducerare (t.ex. rifampicin, dexametason, fenytoin, karbamazepin, rifabutin, rifapentin, fenobarbital och Hypericum perforatum (Johannesört)) kan sänka plasmakoncentrationen av axitinib. Vid val av läkemedel för samtidig administrering rekommenderas sådana utan eller med minimal inducerande effekt på CYP3A4/5. Om en stark CYP3A4/5-inducerare måste ges samtidigt rekommenderas dosjustering av axitinib (se avsnitt 4.2).
In vitro-studier av CYP- och UGT-hämning och -induktion
In vitro-studier visade att axitinib inte hämmar CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 eller UGT1A1 i terapeutiska plasmakoncentrationer.
In vitro-studier visade att axitinib har potential att hämma CYP1A2. Samtidig administrering av axitinib och CYP1A2-substrat kan därför resultera i höjda plasmakoncentrationer av CYP1A2-substrat (t.ex. teofyllin).
In vitro-studier visade också att axitinib har potential att hämma CYP2C8. Samtidig administrering av axitinib och paklitaxel, ett känt CYP2C8-substrat, gav emellertid inte ökade plasmakoncentrationer av paklitaxel hos patienter med framskriden cancer, vilket tyder på att det inte föreligger någon klinisk CYP2C8-hämning.
In vitro-studier på humana hepatocyter visade också att axitinib inte inducerar CYP1A1, CYP1A2 eller CYP3A4/5. Samtidig administrering av axitinib förväntas inte minska plasma-koncentrationen av samtidigt administrerat CYP1A1-, CYP1A2- eller CYP3A4/5-substrat in vivo.
In vitro-studier av P-glykoprotein
In vitro-studier har visat att axitinib hämmar P-glykoprotein. Axitinib förväntas emellertid inte hämma P-glykoprotein vid terapeutiska plasmakoncentrationer. Samtidig administrering av axitinib förväntas därför inte öka plasmakoncentrationen av digoxin, eller andra P-glykoproteinsubstrat, in vivo.
4.6 Fertilitet, graviditet och amning
Graviditet
Det finns inga data från användning av axitinib hos gravida kvinnor. Baserat på axitinibs farmakologiska egenskaper skulle läkemedlet kunna orsaka fosterskador om det ges till en gravid kvinna. Djurstudier har visat reproduktionstoxikologiska effekter, däribland missbildningar (se avsnitt 5.3). Axitinib bör inte användas under graviditet om inte kvinnans kliniska tillstånd kräver behandling med detta läkemedel.
Fertila kvinnor måste använda en effektiv preventivmetod under och upp till en vecka efter avslutad behandling.
Amning
Det är okänt om axitinib utsöndras i bröstmjölk. En risk för det ammade barnet kan inte uteslutas. Axitinib bör inte användas under amning.
Fertilitet
Baserat på icke-kliniska resultat, kan axitinib försämra reproduktionsförmåga och fertilitet hos människa (se avsnitt 5.3).
4.7 Effekter på förmågan att framföra fordon och använda maskiner
Axitinib har mindre effekt på förmågan att framföra fordon och använda maskiner. Patienterna ska informeras om att de kan känna yrsel och/eller trötthet under behandlingen med axitinib.
4.8 Biverkningar
Sammanfattning av säkerhetsprofilen
Följande risker, inklusive lämpliga åtgärder, behandlas mer ingående i avsnitt 4.4: hjärtsviktshändelser, hypertoni, rubbningar i sköldkörtelfunktionen, arteriella tromboemboliska händelser, venösa tromboemboliska händelser, förhöjt hemoglobin- eller hematokritvärde, blödning, gastrointestinal perforation och fistelbildning, sårläkningskomplikationer, PRES, proteinuri samt förhöjda leverenzymvärden.
De vanligaste biverkningarna (≥ 20 %) efter behandling med axitinib var diarré, hypertoni, trötthet, minskad aptit, illamående, viktminskning, dysfoni, palmar-plantar erytrodysestesi (hand-fot-syndrom), blödning, hypotyreos, kräkningar, proteinuri, hosta och förstoppning.
Tabell över biverkningar
I tabell 1 redovisas biverkningar som rapporterades i poolade data från 672 patienter som fick axitinib i kliniska studier av behandling hos patienter med RCC (se avsnitt 5.1). Tabellen inkluderar även biverkningar som identifierats i kliniska studier efter att läkemedlet introducerades på marknaden.
Biverkningarna förtecknas nedan indelade efter organsystem, frekvens och svårighetsgrad. Frekvenskategorierna är: mycket vanliga (≥1/10), vanliga (≥1/100 till <1/10), mindre vanliga (≥1/1 000 till <1/100), sällsynta (≥1/10 000 till <1/1 000), mycket sällsynta (<1/10 000) och ingen känd frekvens (kan inte beräknas från tillgängliga data). Aktuell säkerhetsdatabas för axitinib är för liten för att man ska kunna observera sällsynta och mycket sällsynta biverkningar.
Biverkningarna har placerats i de olika kategorierna på grundval av absolut frekvens i poolade data från kliniska studier. Inom varje organklass redovisas biverkningar med samma frekvens efter fallande svårighetsgrad.
Tabell 1. Biverkningar rapporterade i RCC-studier hos patienter som fick axitinib (n = 672)
|
Systemorganklass |
Frekvens-kategori |
Biverkninga |
Alla graderb % |
Grad 3b % |
Grad 4b % |
|---|---|---|---|---|---|
|
Blodet och lymfsystemet |
Vanliga |
Anemi |
6,3 |
1,2 |
0,4 |
|
Trombocytopeni |
1,6 |
0,1 |
0 |
||
|
Polycytemic |
1,5 |
0,1 |
0 |
||
|
Mindre vanliga |
Neutropeni |
0,3 |
0,1 |
0 |
|
|
Leukopeni |
0,4 |
0 |
0 |
||
|
Endokrina systemet |
Mycket vanliga |
Hypotyreosc |
24,6 |
0,3 |
0 |
|
Mindre vanliga |
Hypertyreosc |
1,6 |
0,1 |
0,1 |
|
|
Metabolism och nutrition |
Mycket vanliga |
Minskad aptit |
39,0 |
3,6 |
0,3 |
|
Vanliga |
Dehydrering |
6,7 |
3,1 |
0,3 |
|
|
Mindre vanliga |
Hyperkalemi |
2,7 |
1,2 |
0,1 |
|
|
Hyperkalcemi |
2,2 |
0,1 |
0,3 |
||
|
Centrala och perifera nervsystemet |
Mycket vanliga |
Huvudvärk |
16,2 |
0,7 |
0 |
|
Dysgeusi |
11,5 |
0 |
0 |
||
|
Vanliga |
Yrsel |
9,1 |
0,6 |
0 |
|
|
Mindre vanliga |
Posteriort reversibelt encefalopatisyndrome |
0,3 |
0,1 |
0 |
|
|
Öron och balansorgan |
Vanliga |
Tinnitus |
3,1 |
0 |
0 |
|
Hjärtat |
Vanliga |
Hjärtsviktshändelserc, d, f |
1,8 |
0,3 |
0,7 |
|
Blodkärl |
Mycket vanliga |
Hypertonig |
51,2 |
22,0 |
1,0 |
|
Blödningc,d, h |
25,7 |
3,0 |
1,0 |
||
|
Vanliga |
Venösa emboliska och trombotiska händelserc,d, i |
2,8 |
0,9 |
1,2 |
|
|
Arteriella emboliska och trombotiska händelserc,d, j |
2,8 |
1,2 |
1,3 |
||
|
Ingen känd frekvens |
Aneurysmer och arteriella dissektionerh |
- |
- |
- |
|
|
Andningsvägar, bröstkorg och mediastinum |
Mycket vanliga |
Dyspnéd |
17,1 |
3,6 |
0,6 |
|
Hosta |
20,4 |
0,6 |
0 |
||
|
Dysfoni |
32,7 |
0 |
0,1 |
||
|
Vanliga |
Orofaryngeal smärta |
7,4 |
0 |
0 |
|
|
Magtarmkanalen |
Mycket vanliga |
Diarré |
55,4 |
10,1 |
0,1 |
|
Kräkningar |
23,7 |
2,7 |
0,1 |
||
|
Illamående |
33,0 |
2,2 |
0,1 |
||
|
Buksmärtor |
14,7 |
2,5 |
0,3 |
||
|
Förstoppning |
20,2 |
1,0 |
0 |
||
|
Stomatit |
15,5 |
1,8 |
0 |
||
|
Dyspepsi |
11,2 |
0,1 |
0 |
||
|
Vanliga |
Smärtor i övre delen av buken |
9,4 |
0,9 |
0 |
|
|
Flatulens |
4,5 |
0 |
0 |
||
|
Hemorrojder |
3,3 |
0 |
0 |
||
|
Glossodyni |
2,8 |
0 |
0 |
||
|
Gastrointestinal perforation och fistelc, k |
1,9 |
0,9 |
0,3 |
||
|
Lever och gallvägar |
Vanliga |
Hyperbilirubinemi |
1,3 |
0,1 |
0,1 |
|
Kolecystitn |
1,0 |
0,6 |
0,1 |
||
|
Hud och subkutan vävnad |
Mycket vanliga |
Palmar-plantar erytrodysestesi (hand-fot-syndrom) |
32,1 |
7,6 |
0 |
|
Utslag |
14,3 |
0,1 |
0 |
||
|
Torr hud |
10,1 |
0,1 |
0 |
||
|
Vanliga |
Pruritus |
6,0 |
0 |
0 |
|
|
Erytem |
3,7 |
0 |
0 |
||
|
Alopeci |
5,7 |
0 |
0 |
||
|
Muskuloskeletala systemet och bindväv |
Mycket vanliga |
Artralgi |
17,7 |
1,9 |
0,3 |
|
Smärtor i extremiteterna |
14,1 |
1,0 |
0,3 |
||
|
Vanlig |
Myalgi |
8,2 |
0,6 |
0,1 |
|
|
Njurar och urinvägar |
Mycket vanliga |
Proteinuril |
21,1 |
4,8 |
0,1 |
|
Vanliga |
Njursviktm |
1,6 |
0,9 |
0,1 |
|
|
Allmänna symtom och/eller symtom vid administreringsstället |
Mycket vanliga |
Trötthet |
45,1 |
10,6 |
0,3 |
|
Astenid |
13,8 |
2,8 |
0,3 |
||
|
Inflammerade slemhinnor |
13,7 |
1,0 |
0 |
||
|
Undersökningar |
Mycket vanliga |
Viktminskning |
32,7 |
4,9 |
0 |
|
Förhöjt lipas |
3,7 |
0,7 |
0,7 |
||
|
Vanliga |
Förhöjt ALAT |
6,5 |
1,2 |
0 |
|
|
Förhöjt amylas |
3,4 |
0,6 |
0,4 |
||
|
Förhöjt ASAT |
6,1 |
1,0 |
0 |
||
|
Förhöjt alkaliskt fosfatas |
4,8 |
0,3 |
0 |
||
|
Förhöjt kreatinin |
5,7 |
0,4 |
0 |
||
|
Förhöjt tyreoideastimulerande hormon |
7,9 |
0 |
0 |
a Biverkningsreaktionerna är ordnade efter med vilken frekvens de uppkommit under behandling, alla orsaker.
b National Cancer Institute Common Terminology Criteria for Adverse Events, Version 3.0
c Se avsnittet ”Beskrivning av ett urval biverkningar” nedan.
d Fatala (grad 5) fall rapporterades.
e Inklusive leukoencefalopati.
f Inklusive hjärtsvikt, kongestiv hjärtsvikt, hjärtlungsvikt, minskad ejektionsfraktion, vänsterkammardysfunktion och högerkammarsvikt.
g Inklusive accelererad hypertoni, förhöjt blodtryck, hypertoni och hypertensiva kriser.
h Inklusive förlängd aktiverad partiell tromboplastintid, blödning från ändtarmen, arteriell blödning, förekomst av blod i urinen, blödning i centrala nervsystemet, cerebral blödning, förlängd koagulationstid, konjunktival blödning, kontusion, blodig diarré, onormal livmoderblödning, epistaxis, ventrikelblödning, gastrointestinal blödning, gingival blödning, hematemes, hematokezi, minskat hematokritvärde, hematom, hematuri, minskat hemoglobinvärde, hemoptys, blödning, blödning från kranskärl, blödning i urinvägarna, hemorrojdblödning, hemostas, ökad benägenhet för blåmärken, förhöjt INR-värde, blödning i de nedre delarna av magtarmkanalen, melena, petekier, faryngeal blödning, förlängd protrombintid, lungblödning, purpura, rektal blödning, minskat antal röda blodkroppar, njurblödning, skleral blödning, hematocele, mjälthematom, splinterblödning, subaraknoidalblödning, blödning från tungan, blödning i de övre delarna av magtarmkanalen och vaginal blödning.
i Inklusive Budd-Chiaris syndrom, djup ventrombos, halsvenstrombos, bäckenventrombos, lungembolism, näthinnevensocklusion, näthinnevenstrombos, ventrombos i nyckelbensvenen, ventrombos och ventrombos i en extremitet.
j Inklusive akut hjärtattack, emboli, hjärtattack, näthinnevensocklusion och transitorisk ischemisk attack.
k Gastrointestinal perforation och fistel inkluderar följande föredragna termer: bukabscess, analabscess, analfistel, fistel, anastomosläckage, gastrointestinal perforation, tjocktarmsperforation, esofagobronkial fistel och peritonit.
l Proteinuri inkluderar följande föredragna termer: äggvita i urinen, förekomst av äggvita i urinen och proteinuri.
m Inklusive akut njursvikt.
n Kolecystit inkluderar akut kolecystit, kolecystit, infektiös kolecystit.
Beskrivning av ett urval biverkningar
Hjärtsviktshändelser (se avsnitt 4.4)
I en kontrollerad klinisk studie av axitinib (n = 359) för behandling av patienter med RCC rapporterades hjärtsviktshändelser hos 1,7 % av de patienter som fick axitinib, däribland hjärtsvikt (0,6 %), hjärtlungsvikt (0,6 %), vänsterkammardysfunktion (0,3 %) och högerkammarsvikt (0,3 %). Hjärtsviktsrelaterade biverkningar av grad 4 rapporterades hos 0,6 % av de patienter som fick axitinib. Hjärtsvikt med dödlig utgång rapporterades hos 0,6 % av de patienter som fick axitinib.
I studier av axitinib som monoterapi (n = 672) för behandling av patienter med RCC rapporterades hjärtsviktshändelser (däribland hjärtsvikt, kongestiv hjärtsvikt, hjärtlungsvikt, vänsterkammardysfunktion, minskad ejektionsfraktion samt högerkammarsvikt) hos 1,8 % av de patienter som fick axitinib. Hjärtsviktshändelser av grad 3/4 rapporterades hos 1,0 % och hjärtsviktshändelser med dödlig utgång rapporterades hos 0,3 % av de patienter som fick axitinib.
Rubbningar i sköldkörtelfunktionen (se avsnitt 4.4)
I en kontrollerad klinisk studie av axitinib för behandling av patienter med RCC rapporterades hypotyreos hos 20,9 % av patienterna och hypertyreos hos 1,1 % av patienterna. Ökning av TSH (tyreoideastimulerande hormon) rapporterades som en biverkning hos 5,3 % av de patienter som fick axitinib. Vid rutinmässiga laboratorieanalyser sågs ökningar av TSH till ≥ 10 μE/ml hos 32,2 % av de patienter vars TSH-värden var < 5 μE/ml före behandlingen.
I poolade kliniska studier med axitinib (n = 672) för behandling av patienter med RCC rapporterades hypotyreos hos 24,6 % av de patienter som fick axitinib. Hypertyreos rapporterades hos 1,6 % av de patienter som fick axitinib.
Venösa emboliska och trombotiska händelser (se avsnitt 4.4)
I en kontrollerad klinisk studie av axitinib för behandling av patienter med RCC rapporterades venösa emboliska och trombotiska händelser hos 3,9 % av de patienter som fick axitinib, inklusive lungemboli (2,2 %), näthinnevensocklusion/-trombos (0,6 %) och djup ventrombos (0,6 %). Venösa emboliska och trombotiska händelser av grad 3/4 rapporterades hos 3,1 % av patienterna som fick axitinib. Fatal lungemboli rapporterades hos en patient (0,3 %) som fick axitinib.
I poolade kliniska studier med axitinib (n = 672) för behandling av patienter med RCC rapporterades venösa emboliska och trombotiska händelser hos 2,8 % av de patienter som fick axitinib. Venösa emboliska och trombotiska händelser av grad 3 rapporterades hos 0,9 % av patienterna. Venösa emboliska och trombotiska händelser av grad 4 rapporterades hos 1,2 % av patienterna. Fatala venösa och trombotiska händelser rapporterades hos 0,1 % av de patienter som fick axitinib.
Arteriella emboliska och trombotiska händelser (se avsnitt 4.4)
I en kontrollerad klinisk studie av axitinib för behandling av patienter med RCC rapporterades
arteriella emboliska och trombotiska händelser hos 4,7 % av patienterna som fick axitinib, däribland hjärtinfarkt (1,4 %), transitorisk ischemisk attack (0,8 %) och cerebrovaskulär händelse (0,6 %). Arteriella emboliska och trombotiska händelser av grad 3/4 rapporterades hos 3,3 % av de patienter som fick axitinib. En fatal akut hjärtinfarkt och en cerebrovaskulär händelse rapporterades hos en patient vardera (0,3 %). I kliniska studier av axitinib som monoterapi (n = 850) rapporterades arteriella emboliska och trombotiska händelser (däribland transitorisk ischemisk attack, hjärtinfarkt och cerebrovaskulär händelse) hos 5,3 % av patienterna som fick axitinib.
I poolade kliniska studier med axitinib (n = 672) för behandling av patienter med RCC rapporterades arteriella emboliska och trombotiska händelser hos 2,8 % av de patienter som fick axitinib. Arteriella emboliska och trombotiska händelser av grad 3 rapporterades hos 1,2 % av patienterna. Arteriella emboliska och trombotiska händelser av grad 4 rapporterades hos 1,3 % av patienterna. Fatala arteriella och trombotiska händelser rapporterades hos 0,3 % av de patienter som fick axitinib.
Polycytemi(se Förhöjt hemoglobin- eller hematokritvärde i avsnitt 4.4)
I en kontrollerad klinisk studie av axitinib för behandling av patienter med RCC rapporterades polycytemi hos 1,4 % av de patienter som fick axitinib. Rutinmässiga laboratorieanalyser detekterade förhöjt hemoglobin som låg över ULN hos 9,7 % av de patienter som fick axitinib. I fyra kliniska studier av axitinib för behandling av patienter med RCC (n = 537) rapporterades förhöjt hemoglobinvärde som låg över ULN hos 13,6 % av de patienter som fick axitinib.
I poolade kliniska studier med axitinib (n = 672) för behandling av patienter med RCC rapporterades polycytemi hos 1,5 % av de patienter som fick axitinib.
Blödning (se avsnitt 4.4)
I en kontrollerad klinisk studie av axitinib för behandling av patienter med RCC, där patienter med obehandlade hjärnmetastaser var exkluderade, rapporterades blödningar hos 10,6 % av de patienter som fick axitinib. Blödningsbiverkningarna hos de patienter som behandlades med axitinib innefattade epistaxis (7,8 %), hematuri (3,6 %), hemoptys (2,5 %), rektal blödning (2,2 %), gingival blödning (1,1 %), ventrikelblödning (0,6 %), cerebral blödning (0,3 %) och blödning i de nedre delarna av magtarmkanalen (0,3 %) . Blödningsbiverkningar av grad ≥ 3 rapporterades hos 3,1 % av de patienter som fick axitinib (däribland cerebral blödning, ventrikelblödning, blödning i de nedre delarna av magtarmkanalen och hemoptys). Fatal blödning rapporterades hos en patient (0,3 %) som fick axitinib (ventrikelblödning). I studier av axitinib som monoterapi (n = 850) rapporterades hemoptys hos 3,9 % av patienterna, hemoptys av grad > 3 rapporterades hos 0,5 % av patienterna.
I poolade kliniska studier med axitinib (n = 672) för behandling av patienter med RCC rapporterades blödning hos 25,7 % av de patienter som fick axitinib. Blödningsbiverkningar av grad 3 rapporterades hos 3 % av patienterna. Blödningsbiverkningar av grad 4 rapporterades hos 1 % av patienterna. Fatal blödning rapporterades hos 0,4 % av de patienter som fick axitinib.
Gastrointestinal perforation och fistelbildning (se avsnitt 4.4)
I en kontrollerad klinisk studie av axitinib för behandling av patienter med RCC rapporterades händelser av typen gastrointestinal perforation hos 1,7 % av patienterna som fick axitinib, inklusive analfistel (0,6 %), fistel (0,3 %) och gastrointestinal perforation (0,3 %). I studier av axitinib som monoterapi (n = 850) rapporterades händelser av typen gastrointestinal perforation hos 1,9 % av patienterna och fatal gastrointestinal perforation hos en patient (0,1 %).
I poolade kliniska studier med axitinib (n = 672) för behandling av patienter med RCC rapporterades gastrointestinal perforation och fistel hos 1,9 % av de patienter som fick axitinib.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning till Läkemedelsverket, men alla kan rapportera misstänkta biverkningar till Läkemedelsverket, www.lakemedelsverket.se. Postadress
Läkemedelsverket
Box 26
751 03 Uppsala
4.9 Överdosering
Det finns ingen specifik behandling vid överdosering av axitinib.
I en kontrollerad klinisk studie av axitinib för behandling av patienter med RCC fick en patient oavsiktligt en dos om 20 mg två gånger dagligen under 4 dagar och kände yrsel (grad 1).
I en klinisk dosundersökningsstudie med axitinib fick de patienter som erhöll startdoser på 10 mg två gånger dagligen eller 20 mg två gånger dagligen biverkningar som hypertoni, kramper kopplade till hypertoni, samt fatal hemoptys.
Vid misstänkt överdosering ska behandlingen med axitinib avbrytas och stödjande behandling sättas in.
5 FARMAKOLOGISKA EGENSKAPER
5.1 Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Antineoplastiska medel, proteinkinashämmare. ATC-kod: L01EK01.
Verkningsmekanism
Axitinib är en potent och selektiv tyrosinkinashämmare för vaskulära endoteliala tillväxtfaktorreceptorer (VEGFR-1, VEGFR-2 och VEGFR-3). Dessa receptorer deltar i den patologiska angiogenesen, tumörtillväxten och metastasutvecklingen vid cancer. Axitinib har visats ha en starkt hämmande effekt på VEGF-medierad endotelcellsproliferation och -överlevnad. Axitinib hämmade fosforyleringen av VEGFR-2 i kärlen i xenografttumörer som uttryckte målproteinet in vivo och resulterade i långsammare tumörtillväxt, tumörregression och hämmad metastasutveckling i flera experimentella cancermodeller.
Effekt på QTc-intervallet
I en randomiserad 2-vägs överkorsningsstudie fick 35 friska försökspersoner en peroral singeldos av axitinib (5 mg) i 7 dagar, med eller utan 400 mg ketokonazol. Resultaten av denna studie visar att en plasmaexponering för axitinib som är upp till dubbelt så hög som den förväntade terapeutiska nivån efter en 5 mg-dos inte medförde någon kliniskt signifikant QT-intervallförlängning.
Klinisk effekt och säkerhet
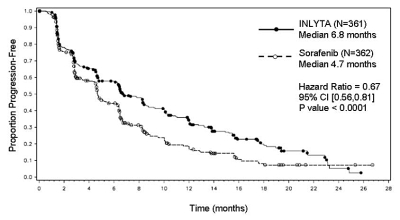
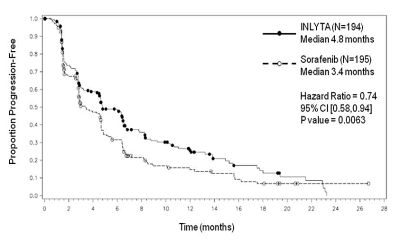
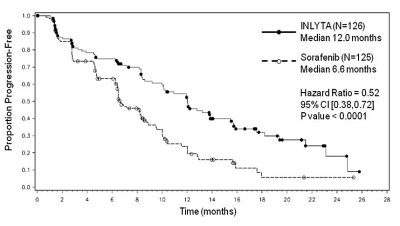
Axitinibs säkerhet och effekt undersöktes i en randomiserad, öppen, multicenterstudie i fas 3. Patienter (n = 723) med framskriden RCC, vars sjukdom hade progredierat under eller efter en tidigare systemisk behandling, däribland sunitinib-, bevacizumab-, temsirolimus- och cytokininnehållande regimer, randomiserades (1:1) till att få axitinib (n = 361) eller sorafenib (n = 362). Det primära effektmåttet var progressionsfri överlevnad (PFS). Utvärdering skedde genom en blindad oberoende central granskning. Sekundära effektmått var objektiv responsfrekvens (ORR) och total överlevnad (OS).
Av de patienter som skrevs in i studien hade 389 (53,8 %) fått en tidigare sunitibbaserad behandling, 251 (34,7 %) hade fått en tidigare cytokinbaserad behandling (interleukin-2 eller interferon-alfa), 59 (8,2 %) hade fått en tidigare bevacizumabbaserad behandling och 24 patienter (3,3 %) hade fått en tidigare temsirolimusbaserad behandling. Demografiska data och sjukdomskaraktäristika vid baslinjen var desamma för axitinib- och sorafenibgrupperna vad avser ålder, kön, ras, ECOG-status (Eastern Cooperative Oncology Group), geografiskt område och tidigare behandling.
I den totala patientpopulationen och i de två huvudsubgrupperna (tidigare behandling med sunitinib respektive tidigare behandling med cytokin) förelåg en statistiskt signifikant fördel för axitinib framför sorafenib avseende det primära effektmåttet PFS (se tabell 2 och figur 1, 2 och 3). Effekten på median-PFS skilde sig åt i subgrupperna, indelade efter typ av tidigare behandling. Två av subgrupperna var för små för att ge tillförlitliga resultat (tidigare behandling med temsirolimus respektive tidigare behandling med bevacizumab). Man såg inga statistiskt signifikanta skillnader mellan grupperna avseende total överlevnad i totalpopulationen eller i subgrupperna, indelade efter tidigare behandling.
Tabell 2. Effektresultat
|
Effektmått/ studiepopulation |
axitinib |
sorafenib |
HR (95 % KI) |
p-värde |
|---|---|---|---|---|
|
ITT totalt |
n = 361 |
n = 362 | ||
|
Median-PFSa,b i månader (95 % KI) |
6,8 (6,4; 8,3) |
4,7 (4,6; 6,3) |
0,67 (0,56; 0,81) |
< 0,0001c |
|
Median OSd i månader (95 % KI) |
20,1 (16,7; 23,4) |
19,2 (17,5; 22,3) |
0,97 (0,80; 1,17) |
NS |
|
ORRb,e % (95 % KI) |
19,4 (15,4; 23,9) |
9,4 (6,6; 12,9) |
2,06f (1,41; 3,00) |
0,0001g |
|
Tidigare sunitinib-behandling |
n = 194 |
n = 195 | ||
|
Median PFS a,b i månader (95 % KI) |
4,8 (4,5; 6,5) |
3,4 (2,8; 4,7) |
0,74 (0,58; 0,94) |
0,0063h |
|
Median OSd i månader (95 % KI) |
15,2 (12,8; 18,3) |
16,5 (13,7; 19,2) |
1,00 (0,78; 1,27) |
NS |
|
ORRb,e % (95 % KI) |
11,3 (7,2; 16,7) |
7.7 (4,4; 12,4) |
1.48f (079; 2,75) |
NS |
|
Tidigarecytokinbehandling |
n = 126 |
n = 126 | ||
|
Median PFS a,b i månader (95 % KI) |
12,0 (10,1; 13,9) |
6,6 (6,4; 8,3) |
0,52 (0,38; 0,72) |
< 0,0001h |
|
Median OS d i månader (95 % KI) |
29,4 (24,5; NE) |
27,8 (23,1; 34,5) |
0,81 (0,56; 1,19) |
NS |
|
ORR b,e % (95 % KI) |
32,5 (24,5; 41,5) |
13,6 (8,1; 20,9) |
2.39f (1,43-3,99) |
0,0002i |
KI = konfidensintervall, HR = riskkvot (axitinib/sorafenib); ITT: Intent-to-treat; NE: kan ej beräknas; NS: ej statistiskt signifikant; ORR: objektiv responsfrekvens; OS: total överlevnad; PFS: progressionsfri överlevnad.
a Tid från randomisering till progression eller död av någon orsak, vilket som inträffar först. Brytdatum: 3 juni 2011.
b Bedömning genom oberoende radiologisk granskning i enlighet med RECIST (Response Evaluation Criteria in Solid Tumours).
c Ensidigt p-värde från ett log-rank test av behandlingen indelad efter ECOG-status och tidigare behandling.
d Brytdatum: 1 november 2011.
e Brytdatum: 31 augusti 2010.
f Riskkvot (RR) används för ORR. Riskkvot > 1 innebär större sannolikhet för respons i axitinib-armen, riskkvot < 1 innebär större sannolikhet för respons i sofarenibarmen.
g Ensidigt p-värde från ett Cochran-Mantel-Haenszel-test av behandlingen indelad efter ECOG-status och tidigare behandling.
h Ensidigt p-värde från ett log-rank test av behandlingen indelad efter ECOG-status.
i Ensidigt p-värde från ett Cochran-Mantel-Haenszel-test av behandlingen indelad efter ECOG-status.
Figur 1. Kaplan-Meier-kurva över progressionsfri överlevnad enligt oberoende bedömning, för totalpopulationen
Figur 2. Kaplan-Meier-kurva över progressionsfri överlevnad enligt oberoende bedömning, för subgruppen tidigare sunitinibbehandling
Figur 3. Kaplan-Meier-kurva över progressionsfri överlevnad enligt oberoende bedömning, för subgruppen tidigare cytokinbehandling
Pediatrisk population
Europeiska läkemedelsmyndigheten har beviljat undantag från kravet att skicka in studieresultat för axitinib för alla grupper av den pediatriska populationen för behandling av cancer i njurar och njurbäcken (exklusive nefroblastom, nefroblastomatos, klarcellssarkom, mesoblastiskt nefrom, medullärt karcinom och rabdoid tumör i njuren) (se avsnitt 4.2 för information om pediatrisk användning).
5.2 Farmakokinetiska egenskaper
Efter oral administrering av axitinibtabletter är den genomsnittliga biotillgängligheten 58 % jämfört med vid intravenös administrering. Axitinibs halveringstid i plasma varierar från 2,5 till 6,1 timmar. När axitinib 5 mg gavs två gånger dagligen resulterade detta i mindre än en dubblering av ackumuleringen jämfört med vid en singeldos. Baserat på axitinibs korta halveringstid förväntas steady state uppnås inom 2–3 dagar efter den första dosen.
Absorption och distribution
Maximal koncentration av axitinib i plasma uppnås vanligen inom 4 timmar efter oral administrering med median Tmax mellan 2,5 och 4,1 timmar. När axitinib administrerades tillsammans med måltid med måttlig fetthalt resulterade det i 10 % lägre exponering jämfört med vid administrering efter nattlig fasta. En måltid med högt fett- och kaloriinnehåll resulterade i 19 % högre exponering jämfört med administrering efter nattlig fasta. Axitinib kan tas med eller utan föda (se avsnitt 4.2).
Genomsnittlig Cmax och AUC ökade proportionellt över doseringsintervallet 5 till 10 mg axitinib. In vitro-bindning av axitinib till humana plasmaproteiner är > 99 %, med preferens för albumin och måttlig bindning till surt alfa-1-glykoprotein. Vid dosen 5 mg två gånger dagligen med måltid, var det geometriska medelvärdet för maximal plasmakoncentration och 24-timmars AUC 27,8 ng/ml respektive 265 ng/ml/tim. hos patienter med avancerad RCC. Geometriskt medelvärde för oral clearance och skenbar distributionsvolym var 38 l/tim. respektive 160 l.
Metabolism och eliminering
Axitinib metaboliseras främst i levern av CYP3A4/5 och i mindre utsträckning av CYP1A2, CYP2C19 och UGT1A1.
Efter oral administrering av en 5 mg radioaktivt axitinibdos återfanns 30 - 60 % av radioaktiviteten i feces och 23 % i urinen. Oförändrat axitinib, motsvarande 12 % av dosen, var den viktigaste komponent som återfanns i feces. Inget oförändrat axitinib återfanns i urinen. Karboxylsyra- och sulfoxidmetaboliter stod för huvuddelen av radioaktiviteten i urinen. I plasma utgjorde N-glukuronidmetaboliten den främsta radioaktiva komponenten (50 % av cirkulerande radioaktivitet) medan oförändrat axitinib och sulfoxidmetaboliten var och en stod för ungefär 20 % av den cirkulerande radioaktiviteten.
Sulfoxid- och N-glukorinidmetaboliterna visar ungefär 400 gånger respektive 8 000 gånger lägre potens mot VEGR-2 in vitro, jämfört med axitinib.
Särskilda populationer
Äldre, kön och etnicitet
Populationskinetiska analyser av patienter med framskriden cancer (däribland avancerad RCC) och friska frivilliga visar att ålder, kön, kroppsvikt, etnicitet, njurfunktion, UTG1A1-genotyp eller CYP2C19-genotyp inte har någon kliniskt relevant effekt.
Pediatrisk population
Axitinib har inte studerats hos barn < 18 år.
Nedsatt leverfunktion
In vitro- och in vivo-data tyder på att axitinib främst metaboliseras i levern.
Vid jämförelse hos patienter med normal leverfunktion var den systemiska exponeringen efter en singeldos av axitinib densamma hos personer med lätt nedsatt leverfunktion (Child-Pugh klass A) och högre (ungefär fördubblad) hos patienter med måttligt nedsatt leverfunktion (Child-Pugh klass B). Axitinib har inte studerats hos personer med allvarligt nedsatt leverfunktion (Child-Pugh klass C) och ska inte användas i denna population (se avsnitt 4.2 för rekommendationer om dosjustering).
Nedsatt njurfunktion
Oförändrat axitinib detekteras inte i urinen.
Axitinib har inte studerats hos personer med nedsatt njurfunktion. I kliniska studier av axitinib för behandling av patienter med RCC, exkluderades patienter med serumkreatinin > 1,5 x ULN eller beräknad kreatininclearance på < 60 ml/min. Populationsfarmakokinetiska analyser har visat att clearance av axitinib inte förändrades hos personer med nedsatt njurfunktion och ingen justering av axitinibdosen krävs.
5.3 Prekliniska säkerhetsuppgifter
Toxicitet vid upprepade doser
De viktigaste toxicitetsreaktionerna hos mus och hund efter upprepad dosering i upp till 9 månader var gastrointestinala, hematopoetiska, reproduktiva, skeletala och dentala, med NOAEL (No Observed Adverse Effect Levels) ungefär motsvarande eller under förväntad exponering hos människa vid den rekommenderade kliniska startdosen (baserat på AUC-nivåerna).
Karcinogenicitet
Karcinogenicitetsstudier med axitinib har inte utförts.
Gentoxicitet
Axitinib var inte mutagent eller klastogent i konventionella gentoxicitetsanalyser in vitro. En signifikant ökning av polyploidi observerades in vitro vid koncentrationer på > 0,22 µg/ml och en ökning av polykromatiska erytrocyter med mikrokärnor sågs in vivo med NOEL (No Observed Effect Level) 69 gånger den förväntade exponeringen hos människa. Resultaten avseende genotoxicitet anses inte kliniskt relevanta vid de exponeringsnivåer som observerats hos människa.
Reproduktionstoxicitet
Axitinibrelaterade fynd i testiklar och bitestiklar är lägre organvikt, atrofi eller degeneration, sänkt antal germinalceller, hypospermi eller onormal spermieform, samt lägre spermietäthet och -antal. Dessa fynd observerades hos mus vid exponeringar på cirka 12 gånger den förväntade exponeringen hos människa, och hos hund vid exponeringar som ligger under den förväntade hos människa. Man såg ingen effekt på parningsförmåga eller fertilitet hos hanmöss vid exponeringar som är cirka 57 gånger den förväntade exponeringen hos människa. Fynd hos honmöss var bl.a. tecken på senare könsmognad, färre eller inga gulkroppar, lägre uterusvikt och uterusatrofi vid exponeringar ungefär motsvarande den förväntade exponeringen hos människa. Nedsatt fertilitet och försämrad embryoöverlevnad observerades hos honmöss vid samtliga testade doser, med exponeringar som vid den lägsta dosen är ungefär 10 gånger den förväntade exponeringen hos människa.
Dräktiga möss som exponerades för axitinib visade en ökad frekvens missbildningar i form av gomspalt och skelettvariationer, däribland fördröjd benbildning, vid exponeringsnivåer som låg under den förväntade hos människa. Inga perinatala och postnatala studier av utvecklingstoxicitet har genomförts.
Toxicitetsfynd hos immatura djur
Reversibel fyseal dysplasi observerades hos mus och hund som fick axitinib under minst en månad vid exponeringsnivåer som var sex gånger högre än den förväntade exponeringen hos människa. Partiellt reversibel tandkaries observerades hos mus som behandlades längre tid än en månad vid exponeringsnivåer motsvarande den förväntade exponeringen hos människa. Andra toxiska reaktioner som kan vara av särskild betydelse för pediatriska patienter har inte utvärderats hos juvenila djur.
6 FARMACEUTISKA UPPGIFTER
6.1 Förteckning över hjälpämnen
Tablettkärna
Mikrokristallin cellulosa
Laktosmonohydrat
Kroskarmellosnatrium
Magnesiumstearat
Filmdragering av tablett
Hypromellos 2910 (15 mPa·s)
Titandioxid (E171)
Laktosmonohydrat
Triacetin (E1518)
Röd järnoxid (E172)
6.2 Inkompatibiliteter
Ej relevant.
6.3 Hållbarhet
3 år.
6.4 Särskilda förvaringsanvisningar
Inga särskilda förvaringsanvisningar.
6.5 Förpackningstyp och innehåll
Inlyta 1 mg filmdragerad tablett
Aluminium-/aluminiumblister som innehåller 14 filmdragerade tabletter. Varje förpackning innehåller 28 eller 56 filmdragerade tabletter.
HDPE-burk med polypropenlock och med kiselgel som torkmedel, innehållande 180 filmdragerade tabletter.
Inlyta 3 mg filmdragerad tablett
Aluminium-/aluminiumblister som innehåller 14 filmdragerade tabletter. Varje förpackning innehåller 28 eller 56 filmdragerade tabletter.
HDPE-burk med polypropenlock och med kiselgel som torkmedel, innehållande 60 filmdragerade tabletter.
Inlyta 5 mg filmdragerad tablett
Aluminium-/aluminiumblister som innehåller 14 filmdragerade tabletter. Varje förpackning innehåller 28 eller 56 filmdragerade tabletter.
HDPE-burk med polypropenlock och med kiselgel som torkmedel, innehållande 60 filmdragerade tabletter.
Inlyta 7 mg filmdragerad tablett
Aluminium-/aluminiumblister som innehåller 14 filmdragerade tabletter. Varje förpackning innehåller 28 eller 56 filmdragerade tabletter.
HDPE-burk med polypropenlock och med kiselgel som torkmedel, innehållande 60 filmdragerade tabletter.
Eventuellt kommer inte alla förpackningsstorlekar att marknadsföras.
6.6 Särskilda anvisningar för destruktion
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
7 INNEHAVARE AV GODKÄNNANDE FÖR FÖRSÄLJNING
Pfizer Europe MA EEIG
Boulevard de la Plaine 17
1050 Bruxelles
Belgien
8 NUMMER PÅ GODKÄNNANDE FÖR FÖRSÄLJNING
Inlyta 1 mg filmdragerade tabletter:
EU/1/12/777/001
EU/1/12/777/002
EU/1/12/777/003
Inlyta 3 mg filmdragerade tabletter:
EU/1/12/777/007
EU/1/12/777/008
EU/1/12/777/009
Inlyta 5 mg filmdragerade tabletter:
EU/1/12/777/004
EU/1/12/777/005
EU/1/12/777/006
Inlyta 7 mg filmdragerade tabletter:
EU/1/12/777/010
EU/1/12/777/011
EU/1/12/777/012
9 DATUM FÖR FÖRSTA GODKÄNNANDE/FÖRNYAT GODKÄNNANDE
Första godkännandet: 03 september 2012
Förnyat godkännande: 22 maj 2018
10 DATUM FÖR ÖVERSYN AV PRODUKTRESUMÉN
07/2021
Ytterligare information om detta läkemedel finns på Europeiska läkemedelsmyndighetens webbplats http://www.ema.europa.eu.